 Revistas
Revistas Archivo
Archivo









































Este artículo es parte de la edición de noviembre, 2021
El mayor estudio de metagenómica realizado nunca revela la enorme complejidad y diversidad de la microbiota intestinal en las aves de producción
Introducción
El tracto gastrointestinal de ave es el hogar de una compleja comunidad de microbios y sus genes – el microbioma -, sustentando los vínculos entre la dieta, la salud y la productividad, como se demuestra por la capacidad de los antibióticos para promover el crecimiento de los pollitos. Esta comunidad microbiana también actúa como fuente de organismos patógenos, causantes de enfermedades en las aves o en el ser humano, además de proporcionar un reservorio de genes de resistencia a los antimicrobianos.
Estudios previos de esta comunidad han documentado una rica variedad de microorganismos – dominado por bacterias, pero incluyendo virus, arqueas y eucariotas microbianos – y han demostrado que la composición taxonómica de la misma varía según la edad, la raza y estado de la enfermedad. Sin embargo, se han basado en análisis de códigos de barras moleculares, que no proporcionan resolución a nivel de especie, son incapaces de detectar virus y no revelan las secuencias del genoma, las estructuras poblacionales o los repertorios funcionales de las especies microbianas.

Para explorar la diversidad en comunidades microbianas complejas hay dos estrategias:
a) un enfoque independiente del cultivo, en base a la secuenciación metagenómica del ADN extraído de muestras relevantes, seguido del perfil de la comunidad según la bioinformática y su análisis y
b) un enfoque dependiente del cultivo, combinando el aislamiento a gran escala de microorganismos en cultivos puros con la secuenciación del genoma completo y su análisis filogenómico.
Para explorar la novedad taxonómica en el microbioma del intestino de pollo, hemos generado los perfiles filogenéticos de una diversidad conocida y desconocida y luego hemos aprovechado los dependientes o no de los cultivos para crear una referencia sin precedentes de una colección de genes microbianos y genomas del intestino de ave de alta calidad, revelando y poniendo nombre a cientos de nuevas especies candidatas de este entorno ecológico, común pero importante.
Resultados
Hemos realizado las secuenciaciones metagenómicas de 90 muestras fecales de aves de dos razas -. Lohman Brown y Sailkie -, tomadas en el sur de Inglaterra, y las analizamos, junto con todos – n = 582 – los metagenomas de ave relevantes de que se dispone públicamente, para agrupar más de 20 millones de genes no redundantes y construir más de 5.500 genomas bacterianos ensamblados con metagenomas. Además, recuperamos casi 600 genomas de bacteriófago, todo lo cual representa la visión más completa hasta la fecha de la diversidad taxonómica dentro del microbioma del intestino aviar, abarcando cientos de nuevos géneros y especies bacterianas candidatas.
Para proporcionar una nomenclatura estable, clara y memorable para especies novedosas, ideamos un sistema combinatorio escalable para la creación de cientos de binomios latinos bien formados. Para ello cultivamos y secuenciamos el genoma de las deyecciones de las aves, documentando más de cuarenta especies novedosas, junto con tres especies del género Escherichia, incluyendo la nueva especie Escherichia whittamii.
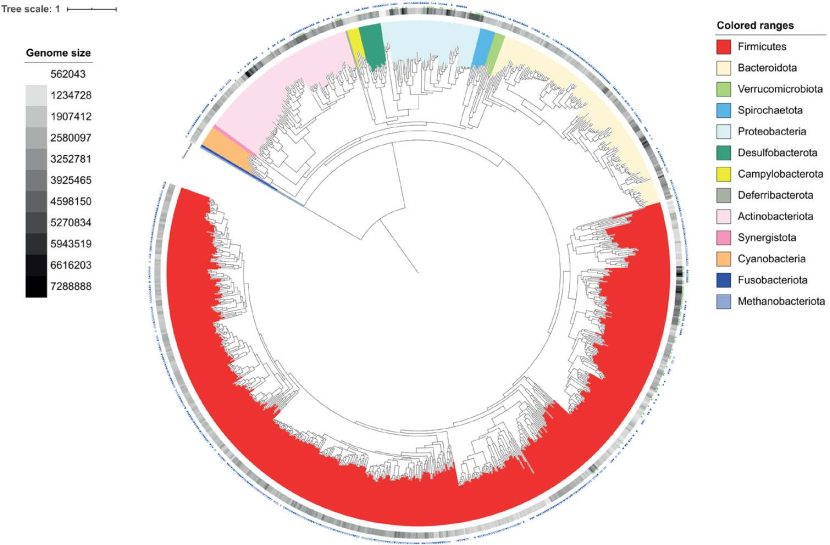
Árbol filogenético de los genomas encontrados en 820 muestras metagenómicas de la flora intestinal y los genomas de 93 especies cultivadas a partir de muestras fecales de aves de producción.
Conclusiones
El extenso catálogo de genes, genomas y aislados que hemos creado mejora sustancialmente los conocimientos sobre el microbioma del intestino de ave en las bases de datos públicas y hace posible secuenciar el perfil del mismo de una forma mucho más rápida, fácil e íntegra, proporcionando un recurso valioso que establece los trabajos para futuro estudios comparativos.
Este estudio también sienta un precedente relevante no solo sobre los microbiomas animales, sino también para estudios sobre todos los microbiomas, asignando binomios latinos bien formados a cientos de especies metagenómicas en una alternativa escalable al uso automatizado de designaciones alfanuméricas sosas, inestables y antipáticas para el usuario. Basándose en el precedente establecido por el estudio actual, recientemente hemos ampliado este enfoque para abarcar la creación de más de un millón de nuevos nombres para bacterias y arqueas. Por lo tanto, ha llegado el momento de traer de nuevo a Linneo al centro de los estudios sobre el microbioma.
RACHEL GILROY Y COL.
PeerJ, DOI 10.7717/peerj.10941
Con el apoyo de:

Categorías
Las seis interprofesionales de la carne se comprometen a que la ganadería española reduzca un 30% sus emisiones
Leer
El sector avícola podría perder más de 300 millones € por incremento de costes de materias primas y subida de la luz
Leer
El aceite de palma y sus ácidos grasos juegan un papel importante en la nutrición animal
Leer
Los avicultores galos denuncian la desinformación de los grupos de presión veganos y animan a los franceses a no dejarse manipular
Leer
Los productores de huevos piden un compromiso social para asumir el coste de criar sin jaulas
Leer
La interprofesional francesa del huevo, alerta ante la prohibición del sacrificio de pollitos
Leer
 PDF
PDF





















































































































